
The question is no longer a debate: scaling up demands new algorithmic strategies. In chemistry, an original approach, the quantum Monte Carlo method or QMC, massively exploits the intrinsic parallelism of probabilistic methods. A savvy implementation can even add real resilience to material failures…
Michel Caffarel, Anthony Scemama
CNRS – Laboratoire de Chimie et Physique Quantiques, Université Paul Sabatier, Toulouse, France.
The availability of massively parallel computers gives rise to new paradigms and opens new routes to numerical simulation. Rather than working hard to parallelize as many algorithms as possible, which do not necessarily lend themselves to the exercise, it can be beneficial to give up the usual approaches and switch to alternative methods. Methods intrinsically inefficient on computers with a few thousand processors but whose algorithmic structure will take advantage of a potentially very large number of processors.
We all know that on a single processor architecture, the optimal algorithm is the one that makes it possible to calculate a given quantity in the smallest possible number of elementary operations (for the sake of simplicity, we’ll ignore possible constraints related to I/O and/or memory limitations). The restitution time (i.e. the time the user waits to obtain the desired result) and the execution time are then essentially identical and proportional to the number of operations to be performed.
In a parallel architecture, the concept of number of operations to be performed becomes secondary, the objective being to reduce the restitution time through parallel calculations, even if this means performing a much larger number of elementary operations. When the restitution time can be made inversely proportional to the number of computation cores used, we are then in a situation of optimal parallelism. When this situation can be extended to arbitrarily large numbers of processors, we can then rightfully speak of methods that scale up.
As supercomputers are bound to come with an ever greater number of processors, having an algorithm that scales up becomes both a real challenge and a reasonable goal. It is the guarantee that there will always be a minimum number of processors for which the algorithm proves superior to any other that does not scale up, whatever its intrinsic performance (single processor).
Virtual Chemistry
The problem is well exemplified in chemistry, a scientific domain extremely demanding in numerical simulations. Advances in drug design, new materials (nanosciences) and renewable energies put it at the heart of our daily lives. Being able to understand, predict and innovate in this field is a considerable society issue.
In parallel with traditional chemistry in the laboratory, a genuine virtual chemistry is developing, where chemical processes are simulated on the computer starting from microscopic equations of matter. With this new “computer chemistry”, scientists seek to reconstruct as faithfully as possible the complex electron exchanges at the origin of chemical bonds, interactions between atoms and finally various desired chemical properties. This problem is a formidable mathematical and data processing challenge since it brings into play the well-known Schrödinger equation of quantum mechanics, whose required solution – the wave function – is a particularly complex function of the set of positions of the atoms’ nuclei and electrons.
During the last fifty years, and always in very close relationship with the development of the hardware and software characteristics of computers, several methods have emerged. From an algorithmic point of view, these are mainly based on iterative schemes for solving very large linear systems requiring vast amounts of calculations with stringent I/O and memory constraints. Unfortunately, because they are based on the processing of very large matrices, these approaches do not support massively parallel computation very well.
Our group is developing an alternative method for chemistry – a method quite different from the usual techniques but that scales up naturally. Relying on an original interpretation of quantum mechanical probabilities, this “Quantum Monte Carlo” (QMC) approach proposes to simulate the real quantum world, where electrons have a delocalized character, by a virtual world where electrons follow classical trajectories like planets around the sun.
In order to introduce the quantum delocalization absent from this scheme, a random component is added to the movement of the electrons. It is this randomness that gives its name to the method: for each electron displacement, random numbers are drawn in a way similar to the Riviera famous casino roulette. Each trajectory or group of random trajectories can be distributed on an arbitrarily large number of computation cores, each one of these trajectories advancing independently of the others (with no communication between them). The situation is thus ideal from the point of view of parallel computation.
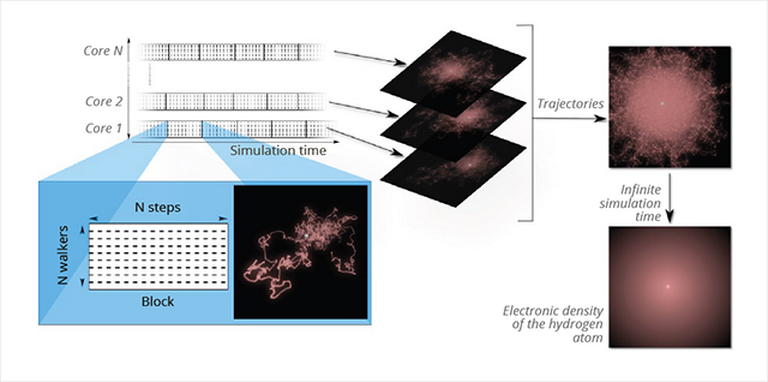
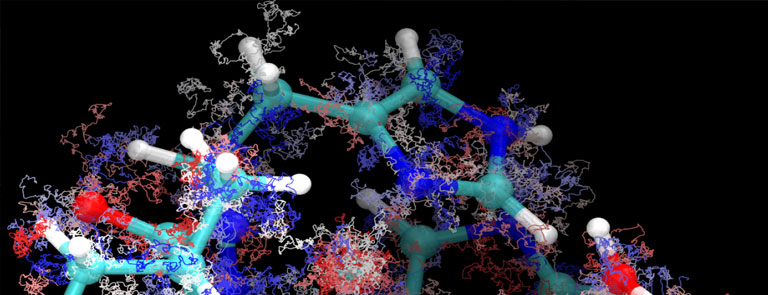
Fig. 1 – Application of the QMC method to the simulation of the hydrogen atom.
Fig. 1 illustrates the QMC method applied to the case of simulating a simple hydrogen atom (one electron moving around a proton). The image inserted in the blue block represents a few thousands steps of a trajectory of the electron moving randomly around the fixed nucleus (in white) of the atom. The trajectories obtained on independent computation cores can be superimposed at will (right part of the figure) and the exact electron density (probability of presence of the electron) is reconstructed in the limit case of an infinite number of trajectories.
More around this topic...








© HPC Today 2024 - All rights reserved.
Thank you for reading HPC Today.


















{kind=link}