
Comment mieux comprendre les mouvements moléculaires qu’en les simulant en temps réel sans contrainte de durée ? C’est à cet objectif, jamais atteint jusqu’ici, que se destine Milestoning, un algorithme capable de reproduire des processus s’échelonnant d’une femtoseconde à plusieurs heures…
Les molécules sont sans doute l’un des éléments les plus “évasifs” de l’univers. Les atomes qui les constituent sont en mouvement pour ainsi dire perpétuel, qui ajustent continuellement leur position sur des échelles de temps atteignant la femtoseconde, soit un quadrillionnième de seconde. Ces transitions atomiques sont à l’origine des structures moléculaires qui actionnent un certain nombre de mouvements biologiques vitaux, comme par exemple l’ouverture ou la fermeture des canaux protéiques régissant le battement cardiaque ou encore le passage d’une molécule à travers une membrane cellulaire.
La technique habituellement utilisée par les laboratoires pour cartographier les étapes d’un mouvement cellulaire consiste à rassembler des milliards de déplacements atomiques mesurables en nanosecondes en une seule image d’une microseconde. Le problème, c’est que pour de nombreux processus biologiques, un intervalle d’une microseconde est tout simplement trop court pour être exploitable scientifiquement. Les ADN polymérases, ces enzymes qui lient l’ADN, prennent par exemple quelques millisecondes pour ajouter un nouveau nucléotide. Quant à la diffusion de certaines substances à travers une membrane cellulaire, elle se déroule parfois sur plusieurs heures. Ces événements ne sont donc tout simplement pas observables en détail sur des prises de vues de quelques microsecondes. Il leur faut une chronométrie adaptée à la variabilité de leur durée.
Au-delà de la milliseconde
C’est pour répondre à ce problème qu’une équipe menée par Ron Elber, Directeur de l’Institute for Computational Engineering and Sciences (ICES) de l’Université du Texas, a développé l’algorithme Milestoning. En découpant le mouvement des molécules en séquences plus courtes puis en réassemblant ces dernières a posteriori, Milestoning est en mesure de réaliser des simulations permettant l’analyse des mouvements cellulaires selon leur propre timing, de l’heure à la milliseconde. “Milestoning permet d’atteindre les échelles de temps qui comptent en biologie et qui pourtant échappent à d’autres méthodes de calcul” confirme Ron Elber. “Cette approche ouvre de nouvelles voies pour l’étude d’un très grand nombre de processus en biosciences.”
Parmi les processus visés figure d’abord le mouvement de la myosine, cette “machine moléculaire” responsable de la contraction et du relâchement des muscles. Un mouvement aussi simple que soulever un bras fait appel au travail commun de milliers de milliards de protéines de myosine. Grâce à Milestoning, l’équipe de Ron Elber prend en compte le mouvement de chaque atome de la protéine et observe son mouvement pendant la milliseconde où elle se contracte. Avec un certain à-propos, le mouvement ressemble à la flexion d’un biceps. “C’est un assemblage de très nombreuses étapes qui mènent à un processus plus lent“, explique Ron Elber en commentant la façon dont les mouvements atomiques fugitifs influencent en définitive le mouvement de la protéine de myosine toute entière.
Séquentialiser le parallélisme
La raison pour laquelle les méthodes conventionnelles ne dépassent pas au mieux l’échelle de la milliseconde dans une simulation telle que celle du cycle de la myosine tient à la différence fondamentale entre l’ordonnancement des étapes qui aboutissent à un mouvement donné et celui des processus typiques des calculateurs actuels. Alors que les étapes biologiques sont séquentielles, les calculs, eux, sont aujourd’hui majoritairement réalisés en parallèle. Rien n’interdit la parallélisation des opérations requises pour simuler une étape d’une femtoseconde. C’est juste que, dans ce type d’approche, les ressources et le temps nécessaires deviennent énormes, d’où la classique limite de la microseconde.
Ron Elber contourne ces difficultés en considérant le processus non comme une ligne de temps continue mais comme un assemblage de points. Un cas classique d’application des techniques d’interpolation. Cette approche considère un certain nombre de situations atomiques sur une période donnée – les bornes ou milestones qui donnent leur nom à la méthode – et applique des principes statistiques et mathématiques pour estimer la trajectoire des atomes dans l’intervalle qui sépare chaque point. Le nombre de bornes varie selon le processus de quelques dizaines à quelques dizaines de milliers, la précision augmentant en même temps que ce nombre.
“C’est un peu comme de lire un gros roman policier. Vous pouvez lire chaque page l’une après l’autre ou tricher en lisant quelques pages vers la moitié du livre et quelques pages à la fin, puis en interpolant ce qui s’est passé au milieu” confie pragmatiquement Ron Elber. La simulation du cycle contraction/relaxation de la myosine a par exemple nécessité 200 bornes, pour le calcul desquelles les ressources du Texas Advanced Computing Center (TACC) ont été sollicitées. C’est d’abord sur le calculateur Lonestar qu’a été préparé l’environnement de simulation, du fait de la présence de nœuds GPU, plus efficaces pour ce job que des nœuds CPU classiques. Ensuite, la majorité des calculs a été réalisée sur Stampede, soit concrètement entre dix et cent mille trajectoires moléculaires possibles entre chaque borne. En pratique, chaque trajectoire individuelle a été “tournée” sur quatre cœurs, mais certaines ont été exécutées sur autant de cœurs que disponibles à un instant T.
Ces calculs du chemin moléculaire entre les bornes ont donc été découpés en segments parallèles, puis leurs résultats réagrégés en fin de course. Les bornes elles-mêmes ont été fixées de façon à isoler les segments du processus moléculaire présentant le plus d’intérêt scientifique, comme par exemple un changement conformationnel, ce qui permet d’augmenter la précision des données dont découlent les hypothèses sur les intervalles.
Aux origines de l’évolution
Pour mettre Milestoning à l’épreuve, Ron Elber l’a confronté à d’autres processus moléculaires se déroulant sur des échelles de temps variées – de la diffusion de molécules à travers une membrane, qui dure plusieurs heures, à l’activation d’enzymes par leur substrat, qui ne prend qu’une microseconde. En filigrane, l’un des objectifs de cette recherche était d’analyser le transport cellulaire en temps réel afin de mieux comprendre le fonctionnement des cellules au début de l’évolution animale. On sait que la membrane de la plupart des cellules récentes est équipée de canaux et de pompes qui permettent le transport de matériaux vers l’intérieur ou l’extérieur, mais il est probable que lors de l’apparition des premières cellules, celles-ci aient été caractérisées par une membrane étanche que les molécules devaient traverser.
Réaction in situ de la relaxation de la myosine interpolée avec 241 autres structures en simulation anatomique avec solvation explicite (image Anthony West et Ron Elber).
Grâce à Milestoning, Ron Elber a pu calculer le temps nécessaire à certaines molécules pour traverser certaines membranes. Par exemple, le sucre passe en quelques microsecondes tandis que les acides aminés – briques constitutives des protéines – ont besoin de plusieurs heures. Or, cet intervalle est trop long pour maintenir le rythme de production protéinique essentiel à la vie de la cellule. La lenteur de cette diffusion suggère qu’il a dû exister aux premiers stades de l’évolution un mécanisme plus efficace pour le passage des acides aminés. “Nous sommes en train de recueillir des données cruciales pour la compréhension de ce mécanisme“, confirme Ron Elber, “mais ces techniques de recherche peuvent également être utilisées hors de la soupe primordiale, notamment pour les besoins plus immédiats de compréhension des mécanismes d’ingestion de différents médicaments par la cellule.”
Confirmation par le vivant
Dans un autre projet de recherche, Ron Elber a travaillé avec Ken Johnson, professeur de biochimie à UT, pour analyser comment les mouvements moléculaires des enzymes transcriptases inverses du VIH déterminent quels nucléotides sont incorporés dans un brin d’ADN. La rétrotranscriptase est en effet essentielle à la survie du VIH dans la mesure où elle utilise le génome ARN natif du virus comme modèle pour la création de copies d’ADN – c’est-à-dire pour la réplication virale. Les mouvements d’enzyme modélisés par Ron Elber ont donné des temps de réaction très similaires à ceux observés au cours des expériences de laboratoire de Johnson, ce qui tend à montrer que les simulations sont correctes. Une meilleure compréhension des conformations moléculaires à chaque étape du processus devrait aider d’autres chercheurs à développer des méthodes d’interruption efficaces. “En principe“, poursuit Ron Elber, “il est possible de perturber la protéine pour qu’elle n’agisse plus à certaines étapes clés. Reste à identifier la méthode la plus facile à interrompre…”
Pour l’heure, les travaux de Ron Elber se focalisent de nouveau sur la myosine. Maintenant qu’on connaît en détail le mouvement de la molécule, Milestoning peut être utilisé pour tester ce qui permettrait de la casser. “On ne sait toujours pas quantifier la force mécanique de la cellule, c’est-à-dire la pression qu’elle est capable de supporter avant de craquer. De la même façon qu’on teste la résistance des éléments d’une machine ou d’une voiture, c’est cette partie de la mécanique cellulaire que l’on tente aujourd’hui d’évaluer“, précise-t-il.
En simulation, l’équipe de Ron Elber a montré qu’une protéine de myosine isolée supporte une pression d’environ 10 à 20 piconewtons mais que, dans le même temps, les protéines de myosine agrégées tiennent des charges beaucoup plus importantes – une observation en ligne avec le principe qui établit à 100 newtons la force nécessaire pour soulever un poids de 10 kilos. Plus précisément, il apparaît que lorsqu’on la sollicite à son maximum, la molécule ne flanche pas. Au contraire, elle semble se renforcer ce qui, en termes moléculaires, correspond à une conformation plus stable. “Effectivement“, confirme Ron Elber, “la molécule devient plus stable lorsqu’on lui fait subir un effort. C’est seulement lorsque celui-ci dépasse un certain seuil qu’on arrive à la casser.” La raison de ce renforcement pourrait être que l’effort supplémentaire rend la molécule plus rigide, ce qui fixe la myosine et lui permet de mieux gérer l’effort. Une hypothèse en cours de validation.
Conçu au départ à Cornell puis développé à l’ICES, Milestoning vient donc compléter une série de codes destinés à l’analyse des phénomènes et les structures moléculaires. La recherche en biologie dépend en effet de plus en plus de ces méthodes de calcul sans lesquelles un nombre croissant de travaux auraient atteint leurs limites pratiques. “Les outils que nous développons on finalement une portée très générale” conclut Ron Elber. “La communauté scientifique peut les utiliser en biologie mais également pour les recherches en événements moléculaires sur des échelles de temps longues.” Il y a fort à parier que cette contribution aujourd’hui mature ouvre des perspectives nouvelles pour un large champ d’applications…
More around this topic...






© HPC Today 2024 - All rights reserved.
Thank you for reading HPC Today.
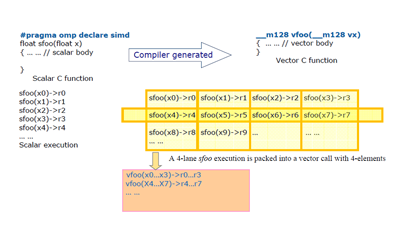

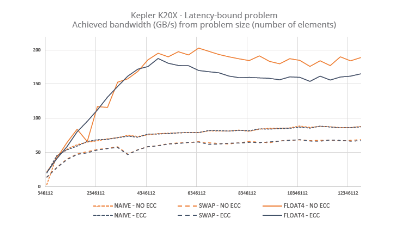






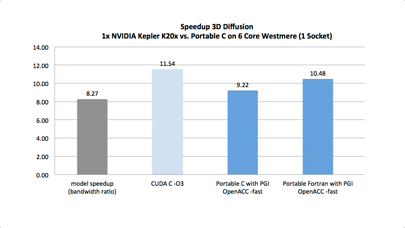











{kind=link}