
La question ne fait maintenant plus débat : le passage à l’échelle commande de nouvelles stratégies algorithmiques. En chimie, une approche originale – la méthode Monte-Carlo quantique ou QMC – exploite massivement le parallélisme intrinsèque des méthodes probabilistes. Une implémentation judicieuse peut même y ajouter une vraie résilience aux pannes matérielles…
Michel Caffarel
Anthony Scemama
CNRS – Laboratoire de Chimie et Physique Quantiques, Université Paul Sabatier, Toulouse
L’apparition récente des calculateurs massivement parallèles donne naissance à un nouveau paradigme et ouvre une voie nouvelle à la simulation numérique. Plutôt que de s’efforcer à paralléliser toujours plus des algorithmes qui ne s’y prêtent pas forcément, il peut être profitable de s’écarter des approches usuelles pour se tourner vers d’autres méthodes. Des méthodes intrinsèquement peu efficaces sur des ordinateurs à quelques milliers de processeurs mais dont la structure algorithmique permet d’exploiter sans difficulté un nombre de processeurs potentiellement très large.
On sait que sur une architecture monoprocesseur, l’algorithme optimal est celui qui permet de calculer une quantité donnée en un nombre d’opérations élémentaires le plus petit possible (par souci de simplicité on ignore ici d’éventuelles contraintes liées aux I/O et/ou à la mémoire centrale). Le temps de restitution, c’est à dire le temps qu’attend l’utilisateur pour obtenir le résultat souhaité, et le temps d’exécution sont alors essentiellement identiques et proportionnels au nombre d’opérations à effectuer.
Dans une architecture parallèle, on sait que la notion de nombre d’opérations à effectuer devient secondaire, l’objectif étant de réduire le temps de restitution grâce au calcul simultané, quitte à effectuer au total un bien plus grand nombre d’opérations élémentaires. Quand le temps de restitution peut être rendu inversement proportionnel au nombre de cœurs de calcul utilisés, on est alors dans une situation de parallélisme optimal. Quand ce régime peut être étendu à des nombres arbitrairement grands de processeurs, on parle alors de méthodes qui “passent à l’échelle”.
Avec le développement des supercalculateurs au nombre de processeurs toujours plus élevé, disposer d’un algorithme qui passe à l’échelle devient donc un véritable défi. C’est la garantie qu’il existera toujours un nombre minimal de processeurs pour lequel l’algorithme sera supérieur à tout autre qui ne passe pas à l’échelle, et cela quelles que soient les performances intrinsèques (monoprocesseur) de ce dernier.
Chimie virtuelle
Cette problématique s’illustre parfaitement dans son application à la chimie, discipline extrêmement gourmande en simulations numériques. La chimie est au cœur de notre vie quotidienne : développement des médicaments (drug design), nouveaux matériaux (nanosciences), énergies nouvelles, etc. Être capable de comprendre, de prédire et d’innover dans ce domaine est un enjeu de société considérable.
En parallèle à la chimie traditionnelle en laboratoire se développe une véritable chimie virtuelle où les processus sont simulés sur ordinateur à partir des équations microscopiques de la matière. Avec cette nouvelle “chimie sur ordinateur”, les scientifiques cherchent à reconstituer le plus fidèlement possible les échanges électroniques complexes à l’origine des liaisons chimiques, des interactions entre atomes et finalement des diverses propriétés chimiques recherchées. Ce problème constitue un formidable défi mathématique et informatique puisqu’il met en jeu la célèbre équation de Schrödinger de la mécanique quantique dont la solution recherchée – la fonction d’onde – est une fonction particulièrement complexe de l’ensemble des positions de tous les électrons (des milliers, voire plus !) et des noyaux des atomes.
Durant ces cinquante dernières années, et toujours en relation très étroite avec le développement des caractéristiques matérielles et logicielles des ordinateurs, plusieurs méthodes ont émergé. D’un point de vue algorithmique, elles s’appuient principalement sur des schémas itératifs de résolution de systèmes linéaires de très grandes tailles nécessitant des volumes de calculs et des besoins en I/O et/ou en mémoire centrale considérables. Malheureusement, parce qu’elles sont basées sur la manipulation et le traitement de très grandes matrices, ces approches ne se prêtent pas facilement au calcul massivement parallèle.
Nous développons dans notre groupe une approche alternative pour la chimie, très différente des approches usuelles mais qui passe à l’échelle naturellement. Basée sur une interprétation originale des probabilités de la mécanique quantique, cette approche connue sous le nom de “méthode Monte-Carlo quantique” (quantum Monte-Carlo ou QMC) propose de simuler le monde quantique réel, où les électrons possèdent un caractère délocalisé, par un monde fictif où les électrons suivent des trajectoires “classiques” comme les planètes autour du soleil.
Afin d’introduire la délocalisation quantique absente de ce schéma, une composante aléatoire est ajoutée au mouvement des électrons. C’est ce caractère aléatoire qui donne son nom à la méthode : à chaque déplacement d’électron un tirage au sort est effectué comme à la roulette du casino de la fameuse station balnéaire. Chaque trajectoire ou groupe de trajectoires aléatoires peut être distribué à volonté sur un nombre arbitrairement grand de cœurs de calcul, chacune de ces trajectoires évoluant indépendamment des autres (pas de communications entre elles). La situation est donc idéale du point de vue du calcul parallèle.
La Figure 1 illustre la méthode QMC appliquée au cas de la simulation de l’atome d’hydrogène, le plus simple des atomes (un électron “gravitant” autour d’un proton). L’image insérée dans le bloc bleu représente quelques milliers de pas d’une trajectoire de l’électron se déplaçant aléatoirement autour du noyau fixe (en blanc) de l’atome. Les trajectoires obtenues sur des cœurs de calcul indépendants peuvent être superposées à volonté (partie droite de la figure) et la densité électronique exacte (probabilité de présence de l’électron) est reconstruite dans le cas-limite d’un nombre infini de trajectoires.
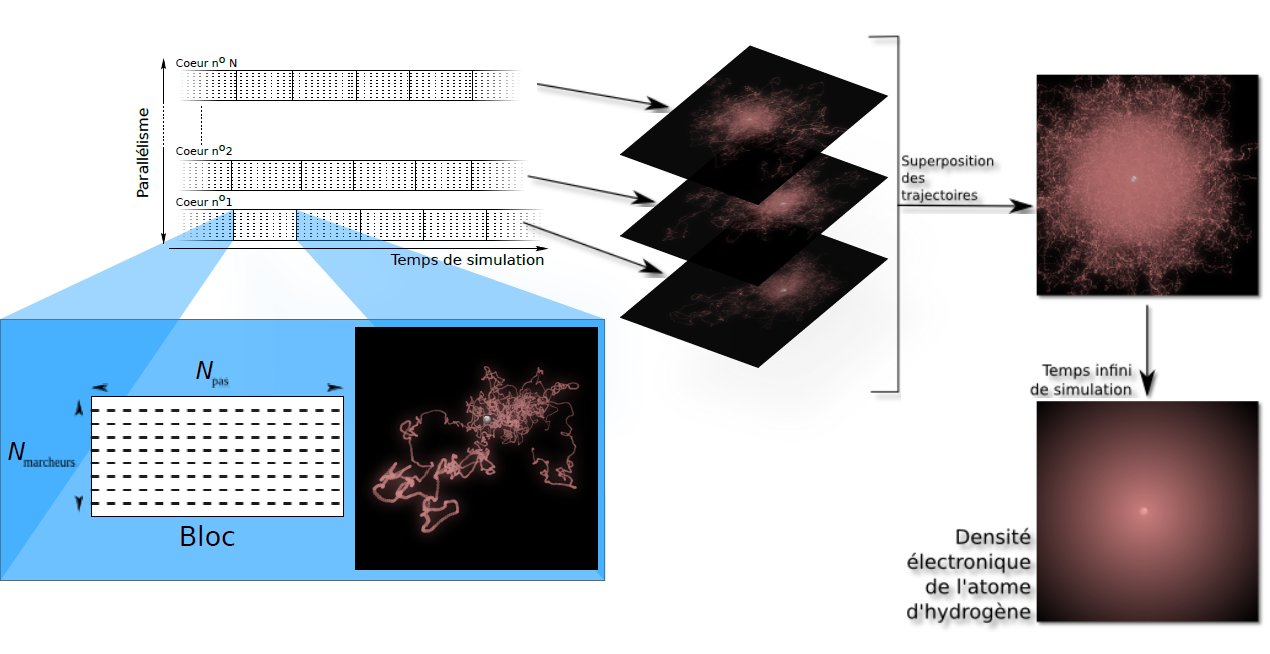
Fig. 1 : Application de la méthode QMC à la simulation de l’atome d’hydrogène.
Aspects computationnels
Déployer efficacement des simulations où des milliers d’électrons effectuent des milliards de pas nécessite d’optimiser au mieux les aspects algorithmiques et informatiques les plus critiques. Détaillons maintenant les principales stratégies que nous avons employées.
Nous définissons un marcheur comme l’ensemble des coordonnées x, y et z de chacun des N électrons du système (un vecteur à 3 N dimensions). Pour réaliser une trajectoire, un marcheur va effectuer une marche aléatoire dans l’espace à 3 N dimensions, de façon à reconstruire la densité de probabilité correspondant au carré de la fonction d’onde.
Nous définissons un bloc (Figure 1) comme un ensemble de trajectoires courtes réalisées par un groupe de marcheurs pendant un certain nombre de pas (de l’ordre de 10 000). Pour chaque bloc, on calcule les valeurs moyennes des propriétés (énergie, moment dipolaire, etc.) à partir des positions successives de chacun des marcheurs le long de toutes les trajectoires du bloc. Si les blocs sont suffisamment longs, les positions finales des marcheurs sont complètement décorrélées des positions initiales, de sorte qu’on peut considérer les blocs comme étant tous indépendants. Dans ce cas, les valeurs moyennes calculées sur les blocs ont une distribution statistique obéissant à une loi Gaussienne. Chaque moyenne de bloc est un échantillon que l’on utilise pour calculer statistiquement les valeurs moyennes des propriétés.
Le but est alors de calculer le plus de blocs possible le plus rapidement possible. Pour cela nous avons adopté deux stratégies. La première est la mise en place d’un système de parallélisation efficace. La seconde est la réduction du temps de calcul de chaque bloc en travaillant sur l’optimisation mono-cœur.
Gérer la résilience
Lorsque l’on utilise des dizaines de milliers de cœurs, une nouvelle préoccupation devient la tolérance aux pannes. En effet, si un nœud de calcul tombe en panne en moyenne au bout de cinq ans, un calcul utilisant simultanément 2 000 nœuds ne pourra pas atteindre 24 heures de calcul sans défaillance. Nous devons donc disposer d’un système qui survive coûte que coûte.
Dans le cas d’algorithmes déterministes le calcul est découpé en tâches, l’accomplissement de chacune d’elles étant indispensable pour obtenir un résultat correct. En cas de panne, certaines tâches ne pourront pas être effectuées.
Fig. 2 : Les clients sont organisés en arbre binaire afin d’alléger les communications réseau : un client envoie son tampon de résultats à son parent dans l’arbre. Lorsque le parent reçoit des données provenant d’un de ses fils, il les ajoute à son tampon de résultats, qui sera envoyé plus tard à son parent (le grand-père), etc. Ainsi, le serveur reçoit par le réseau un petit nombre de gros paquets de résultats au lieu d’un grand nombre de petits paquets. Résilience : si un parent ne répond pas, le client va envoyer son tampon de résultats à son grand-père. Si le grand-père ne répond pas non-plus, le client va l’envoyer à l’arrière grand-père, etc., jusqu’à ce que le paquet finisse par être directement envoyé au serveur de données.
Les bibliothèques MPI sont bien adaptées aux calculs déterministes. En effet, lorsqu’un client MPI ne répond plus, tout le reste de la simulation est tué car certaines tâches ne pourront pas être réalisées. Dans notre modèle, perdre des blocs au cours de la simulation ne change pas les moyennes, mais influe seulement sur la barre d’erreur du résultat. Nous avons donc choisi de ne pas utiliser MPI pour la parallélisation, mais nous avons implémenté un modèle client/serveur, où les clients calculent des blocs et envoient les résultats au serveur qui les stocke dans une base de données. Si un client meurt, tant pis, les autres clients continuent à calculer.
Les clients et les serveurs sont des scripts Python multithreadés qui communiquent par sockets TCP (Figure 3). Notons que les communications étant non-bloquantes et peu intensives, les latences dues à la couche TCP ne posent pas de problème. Il y a un seul client par nœud de calcul. Chaque client lance autant de programmes de calcul qu’il y a de cœurs physiques sur le nœud. Le programme de calcul est un binaire Fortran monothread connecté au client par un pipe Unix. Dès que le résultat est communiqué au client, le programme de calcul commence immédiatement à calculer le bloc suivant. Simultanément, lorsque le client est inactif il transmet ses résultats à un autre client dans le but de les acheminer vers le serveur.
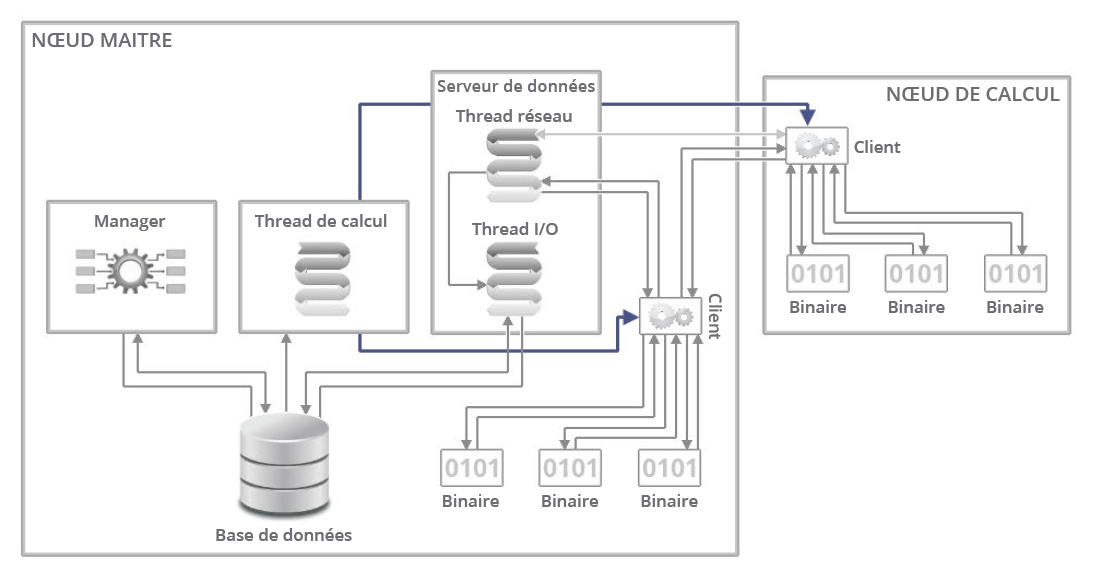
Fig. 3 : Schéma fonctionnel du modèle algorithmique client-serveur.
Le serveur comporte deux threads principales : une thread réseau et une thread I/O. Le serveur reçoit des données par la thread réseau, les traite et les met dans une file d’attente. Simultanément la thread I/O vide la file d’attente pour mettre les résultats sur disque. Les clients n’accèdent jamais au disque dur local ou au système de fichiers partagé mais plutôt à un disque virtuel en RAM (/dev/shm) afin d’échapper aux pannes liées au stockage temporaire (disque plein, panne physique, etc.).
Facteur d’accélération en fonction du nombre de nœuds
Puisque les blocs sont indépendants, aucune synchronisation n’est nécessaire entre les clients, et cela permet d’obtenir un facteur d’accélération presque idéal en fonction du nombre de nœuds. Cependant, il y a deux étapes critiques qui vont nuire à l’idéalité du facteur d’accélération : l’initialisation et la terminaison.
Pour avoir une initialisation rapide, il faut démarrer les clients aussi vite que possible sur les nœuds de calcul. La méthode qui nous paraît la plus rapide est l’utilisation d’un lanceur MPI qui va envoyer par MPI_broadcast un fichier tar contenant le binaire statique Fortran, les scripts Python et le fichier d’input sur tous les nœuds. Lorsqu’un nœud reçoit l’archive, il la décompresse et lance le client qui démarre immédiatement les programmes de calcul. Lorsque tous les clients ont démarré, le lanceur MPI se termine. Ainsi, chaque nœud de calcul démarre dès que possible. Pour un calcul sur un millier de nœuds, nous avons mesuré un temps d’initialisation de l’ordre d’une vingtaine de secondes.
Pour la terminaison, sachant que tous les nœuds de calcul sont désynchronisés, il faudrait attendre que chacun des nœuds ait fini de calculer son bloc. Dans ce cas, il peut en résulter un temps d’attente important où beaucoup de nœuds attendent que les derniers aient terminé. On ne peut pas non plus tuer violemment tous les clients, car les résultats des blocs en cours seraient perdus.
Nous avons donc permis aux programmes de calcul d’intercepter le signal SIGTERM afin d’écourter le bloc en cours et de transmettre le résultat de ce bloc au client. Ainsi, la terminaison d’un client est presque immédiate et aucune seconde de calcul n’est perdue. Pour un calcul d’un millier de nœuds, nous avons mesuré une terminaison de l’ordre de 10 secondes.
Tous ces éléments combinés permettent d’obtenir la courbe de la Figure 4. À nombre constant de nœuds, le temps d’initialisation et de terminaison est constant, donc plus le temps de calcul est long, plus l’efficacité parallèle est bonne.
Optimisations mono-cœur
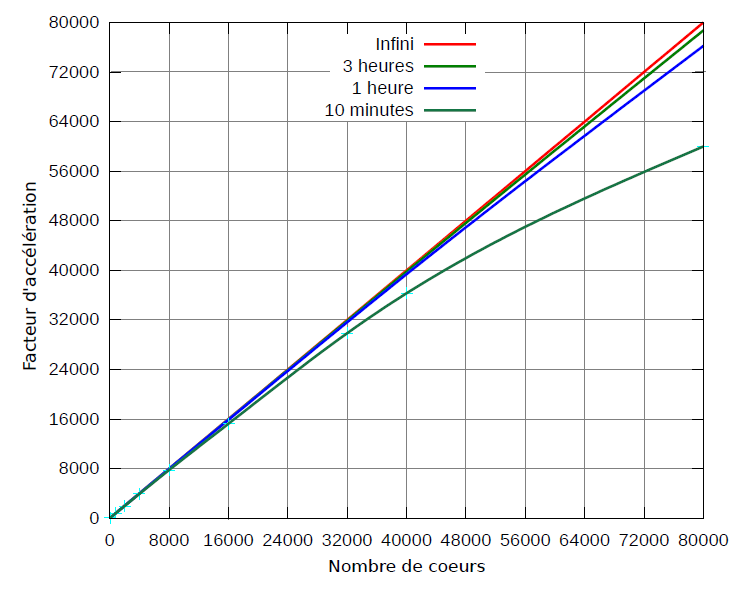
Fig. 4 : Mesure de l’efficacité parallèle résultant des différentes stratégies combinées.
Chaque seconde gagnée dans le programme de calcul se répercutera sur la totalité de la simulation car aucune synchronisation n’est bloquante. Ainsi, nous avons entrepris un travail important d’optimisation mono-cœur de notre programme QMC=Chem en profitant de l’expérience et des outils développés par le groupe du Pr Jalby au Laboratoire Exascale Computing Research (Intel-CEA-UVSQ-GENCI) à l’Université de Versailles-Saint-Quentin-En-Yvelines.
Dans notre algorithme, nous devons évaluer à chaque pas Monte-Carlo (des milliards de pas sont effectués !) la fonction d’onde, ses dérivées par rapport à chacune des coordonnées électroniques et son Laplacien. Ces opérations font intervenir des produits de petites matrices (< 1000 x 1000) dont l'une est dense et l'autre est creuse.
L’outil d’analyse statique MAQAO développé par nos collègues de Versailles nous a aidé à écrire une routine de produit matrice dense x vecteur creux dont les boucles les plus internes atteignent théoriquement les 16 flops/cycle en simple précision sur les processeurs Intel Sandy Bridge du calculateur pétaflopique CURIE (TGCC-GENCI-CEA). Pour cela, nous avons effectué les modifications suivantes dans le but de favoriser le plus possible la vectorisation :
– Tous les accès à la mémoire sont consécutifs
– Tous les tableaux sont alignés sur 32 octets en utilisant des directives du compilateur
– La dimension la plus interne des tableaux multidimensionnels est toujours un multiple de 8 éléments, afin que chaque colonne du tableau soit alignée sur 32 octets
– Déroulage de la boucle externe (unroll and jam) pour réduire le nombre d’écritures en mémoire
– Distribution des boucles pour ne pas dépasser les 16 registres et donc réduire les accès au cache L1.
En pratique, étant donné que nous faisons de l’ordre de N² opérations (N, nombre d’électrons) pour N² accès à la mémoire, le calcul est inévitablement limité par les accès à la mémoire et nous avons mesuré jusqu’à 61% de la performance crête du processeur dans cette routine de produit matrice x vecteur.
Lors de l’installation de CURIE en décembre 2011 et grâce au soutien des ingénieurs de BULL, constructeur de la machine, nous avons pu réaliser notre tout premier calcul QMC grandeur nature sur l’ensemble des ≈ 80 000 cœurs de la machine. La machine étant en cours d’installation, les calculs ont subi des interruptions et des pannes. Ce qui initialement nous avait apparu comme une gêne est devenu finalement une chance : le fait que nos simulations aient pu être menées à bien malgré ces difficultés a validé en pratique la grande robustesse de notre schéma.
Simulations pétaflopiques pour la chimie d’Alzheimer
Grâce aux moyens exceptionnels mis à notre disposition ces deux dernières années par GENCI et le consortium européen PRACE, notre groupe a pu démontrer la faisabilité pratique de nos simulations pour des systèmes difficiles à atteindre par les méthodes usuelles du domaine. Les applications abordées ont concerné la chimie de la maladie d’Alzheimer qui met en jeu des interactions particulièrement délicates à reproduire.
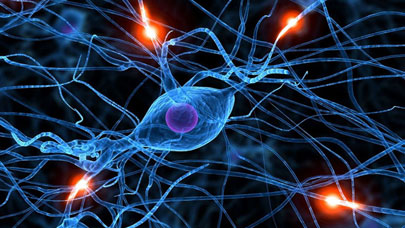
On sait que cette maladie est associée à une dégénérescence du cerveau liée à l’apparition de plaques composées d’agrégats de molécules (peptides) dites β-amyloïde. Comprendre pourquoi et comment ces molécules s’agrègent est un des enjeux majeurs de la compréhension des mécanismes les plus fondamentaux à l’origine de l’apparition de cette maladie grave. En collaboration avec le groupe de biochimie expérimentale du Pr Faller (LCC, Toulouse), nous avons pu démontrer que la précision chimique nécessaire pour décrire quantitativement ces systèmes pouvait être atteinte en utilisant le calculateur CURIE. L’image qui ouvre cet article illustre comment les trajectoires aléatoires des électrons se déploient dans le cas des systèmes chimiques simulés.
Lors de ces simulations, un débit de calcul réel de presque 1 Pétaflops soutenu pendant plusieurs heures (précisément, 960 Tflops/s en simple et double précision) a pu être mesuré.
Même si beaucoup reste à faire, nos premières simulations probabilistes sur une architecture massivement parallèle ouvrent des perspectives prometteuses. Bien que les volumes de calcul nécessaires – des millions d’heures CPU monoprocesseur – soient encore beaucoup trop importants pour en faire des méthodes de routine, l’arrivée d’ici 2020 des machines exaflopiques mille fois plus puissantes permet d’espérer transformer ces techniques émergentes en un outil de simulation accessible à la communauté scientifique la plus large.
More around this topic...






© HPC Today 2024 - All rights reserved.
Thank you for reading HPC Today.


















{kind=link}