Acceleration factor according to the number of nodes
Since blocks are independent, no synchronization is necessary between clients, which gives an almost ideal acceleration factor according to the number of nodes. However, two critical stages can harm the acceleration factor: initialization and termination.
For a fast initialization, it is necessary to start the clients as quickly as possible on the computation nodes. The method that proved the fastest is the use of an MPI launcher that will send by MPI broadcast a tar file containing the Fortran static binary, the Python scripts and the input file on all the nodes. When a node receives the file, it decompresses it and launches the client which immediately starts the calculation programs. When all clients have started, the MPI launcher terminates. Thus, each computation node starts as soon as possible. For a calculation on a thousand nodes, we have measured an initialization time of about twenty seconds.
For termination, taking into account that all the computation nodes are desynchronized, it would be expected that each node has finished calculating its block. This would imply a significant waiting time (many nodes waiting for the last one to finish). We cannot kill all clients either – the results of the blocks in progress would be lost.
We have therefore allowed the calculation programs to intercept the SIGTERM signal in order to cut short the block in progress and to transmit the result of this block to the client. Thus, the termination of a client is almost immediate and no calculation second is lost. For a thousand node calculation, we measured a termination time of about 10 seconds.
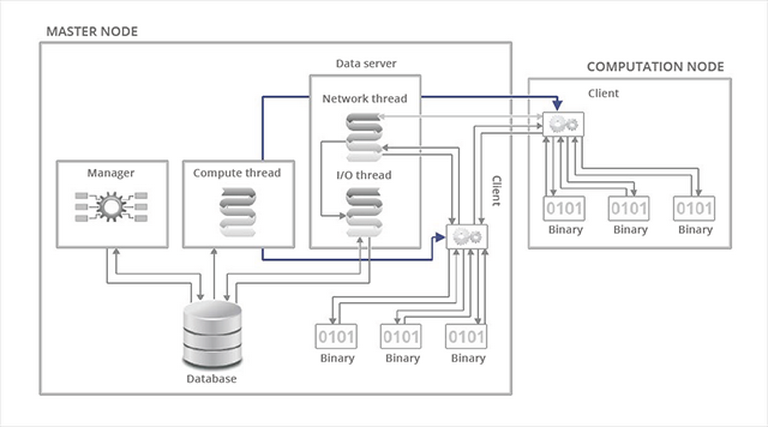
Fig. 3 – Functional diagram of the algorithmic client-server model.
All these elements combined yield the curve of Fig. 3. With a constant number of nodes, initialization and termination times are constant. In other words, the longer the calculation time, the better the parallel efficiency.
Single core optimizations
Each second gained in the calculation program will have an effect on the totality of the simulation, because no synchronization is blocking. With this in mind, we have undertaken an important single core optimization work on our QMC=Chem program, benefiting from the experience and tools developed by Prof. Jalby’s group at Intel’s Exascale Computing Research Laboratory (Intel-CEA-UVSQ-GENCI) at the University of Versailles-Saint-Quentin-En-Yvelines in France.
In our algorithm, at each Monte Carlo step (billions of which are carried out), we must evaluate the wave function, its derivatives with respect to each of the electron coordinates and its Laplacian. These operations use products of small matrices (< 1000 x 1000), of which one is dense and the other is sparse. The MAQAO statistical analysis tool developed by our Versailles colleagues helped us write a dense matrix x sparse vector routine whose innermost loops theoretically reach 16 flops/cycle in single precision on the Intel Sandy Bridge CPUs of the French CURIE Pflops computer (CEA-TGCC-GENCI). For that, we have made the following modifications with the purpose of favoring vectorization as much as possible:
– memory accesses are consecutive;
– arrays are 32-bytes aligned, using compiler directives;
– the innermost dimension of the multidimensional arrays is always a multiple of 8 elements, so that each column of the array is 32-bytes aligned;
– the external loop is unrolled (unroll and jam) to reduce the number of memory stores;
– distribution of the loops does not exceed 16 registers, thus reducing accesses to the L1 cache.
In practice, since we do about N² operations (N, number of electrons) for N² memory accesses, the calculation is inevitably limited by these memory accesses. We have measured up to 61% of peak processor performance in this matrix x vector product routine.
When CURIE was installed in December 2011, we have been able, thanks to the support of the engineers at BULL, the manufacturer of the machine, to carry out our very first large-scale QMC calculation on the approximately 80,000 cores of the machine. As is often the case with a computer being set up, the calculations underwent interruptions and failures. But what first was a trouble to us eventually proved to be an opportunity: the fact that our simulations went through in spite of these difficulties validated practically the robustness of our scheme.
More around this topic...








© HPC Today 2024 - All rights reserved.
Thank you for reading HPC Today.